Phosphorylation - the PTM of choice for cell signalling
Phosphorylation [has been termed](http://dx.doi.org/10.1042/BJ20131014) 'the PTM of choice' for transferring cellular information (cell signalling). Specifically, phosphorylation provides rapid and accurate transmission from cell surface receptors to the nucleus, through [either](http://dx.doi.org/10.1016/S0960-9822(95)00151-5) regulated translocation to the nucleus ([nucleocytoplasmic shuttling](http://dx.doi.org/10.1016/S0014-5793(01)02487-5)) where for example a kinase may in turn phosphorylate transcription factors, or through regulated activation in the cytoplasm. A fair example of such a kinase-TF pair is PKA and CREB, 'transducing' the build-up of intracellular cAMP through its binding to PKA, liberating the kinase's catalytic subunit to move into the nucleus. Its phosphorylation of CRE-binding proteins (CREB) enables them to bind upstream of CRE-response elements on genomic DNA, activating the cAMP-responsive gene(s) downstream. Another more complex mechanism plays out in mitogen-activated protein kinase (MAPK) cascades, induced by a variety of extracellular signals such as mitogens (growth signals), cell-cell interactions, osmotic stress, UV irradiation, and other stresses. The external stimulus precipitates activation of the cascade's kinases in turn, and in some cases ensures high specificity through the involvement of [scaffolding proteins](http://en.wikipedia.org/wiki/Scaffold_protein), *e.g.* [Ste5](http://dx.doi.org/10.1016/0092-8674(94)90427-8) (it's [uncertain](http://dx.doi.org/10.1016/j.tibs.2009.06.007) whether scaffolds are a general phenomenon, due to the poor characterisation of many kinases). The ratios of amino acid targetting by kinases are [on the order of](http://dx.doi.org/10.1098%2Frstb.1998.0228) 1000:100:1 for the O-phosphates of pSer, pThr, and pTyr respectively — a figure [widely](http://dx.doi.org/10.1021%2Fpr900069n) [reported](http://dx.doi.org/10.1007/s10540-005-2846-0) from ~90%/5%/0.05% proportions in chicken cells (pTyr was in fact a serendipitous discovery from studies of Rous sarcoma virus). 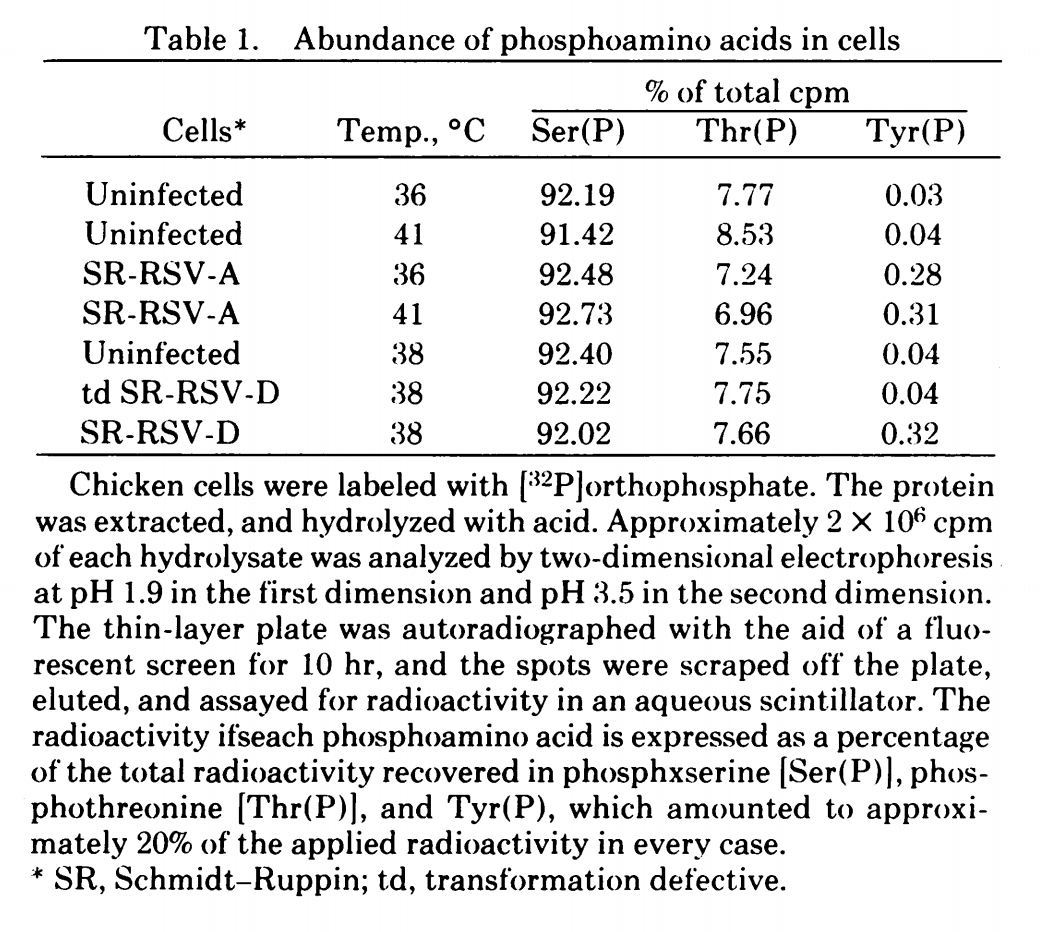
Tony Hunter (director of the Salk Institute Cancer Research Center) has noted that the N-phosphate pHis is 10-100-fold more abundant than pTyr (elsewhere some papers wrongly pigeonhole it with pCys as 'infrequently used'). Histidine phosphorylation forms a regulatory mechanism in higher eukarya, as well as a phosphate transfer intermediate, but is chemically unstable, with a relatively short life in aqueous solution at physiological pH.
Hunter's 1997 Croonian Lecture given to the Royal Society noted that over a third of proteins can be phosphorylated in a mammalian cell, and up to 5% of vertebrate genes will encode kinases or their counterpart phosphatases (the kinase complement in humans is said to be ~2%). The use of phosphotyrosine in such rare cases was, in his words, "not immediately obvious".
> Although P.Tyr is as stable chemically as P.Ser and P.Thr at physiological pH, the turnover numbers for PTPs [protein tyrosine phosphatases] are very high, and this property, in combination with the generally tight negative regulation of PTK [protein tyrosine kinase] activity, means that most PTK substrates have a very low basal level of tyrosine phosphorylation, which is a critical requirement for an inducible signalling system with maximal gain. One plausible reason for the adoption of tyrosine phosphorylation for intercellular signalling was the successful evolution of high affinity P.Tyr-binding domains, such as the SH2 domain, which recognize P.Tyr in a defined sequence context and thereby facilitate specific inducible protein-protein interactions in signalling pathways. Phosphorylated tyrosine may have been selected for this purpose because the aromatic side chain of P.Tyr affords significantly greater binding energy than the aliphatic side chain of P.Ser or P.Thr.
Signalling PTKs target either enzymes, adaptors, docking proteins, or structural proteins to transmit messages. Enzymes and adaptors comprise a modular series of discrete domains, many with the aforementioned SH2 or SH3 domains. These domains mediate interaction with phosphorylation sites - one class of "linear motif" recognition sequence which are enriched within intrinsically disordered regions (IDRs) of cellular proteins.
Such motifs are said to be 'complex promoting', and create low affinity interactions with binding partners, producing signal transduction in a combinatorial manner. They may occur many times over the length of a protein, promoting high avidity interactions, and the recruitment of multiple factors such as is seen in T-cell receptor signalling's LAT complex. In addition to activating, or 'switching on' like this, phosphorylation-induced complexes through these motifs can 'switch away' from other signalling pathways, where linear motifs overlap: mutually exclusive binding vs. LAT's cooperative binding (of SH2- and SH3 domain-binding pTyr motifs).
Ser/Thr-phosphorylated enzymes tend to be found in the intracellular milieu, whereas Tyr-phosphorylated ones are primarily transmembrane.
Phosphorylation was selected for versatility and reversibility
Phosphates are predominantly dianionic at physiological intracellular pH (~7), with the phosphate [mono]ester groups found on proteins bearing one full negative charge and another either partial or full dependent on chemical context. In 2012, Hunter contributed an excellent review to an issue of Philosophical Transactions of the Royal Society B, which he'd curated around the theme of protein phosphorylation's evolution along with Tony Pawson.
> The unique size of the ionic shell [bearing 5 valence shell electrons, and subsequently the large size of the hydration shell] and charge properties of covalently attached phosphate allow specific and inducible recognition of phosphoproteins by phosphospecific-binding domains in other proteins, thus promoting inducible protein–protein interaction. In this manner, phosphorylation serves as a switch that allows signal transduction networks to transmit signals in response to extracellular stimuli.
> …Importantly, when two nucleosides are linked by a phosphate diester, the phosphate is still fully ionized (pKa −. Any molecule with a phosphate ester, including phosphorylated proteins, has these same attributes. The enormous stability of phosphate esters in water at pH 7 (phosphate monoesters are estimated to have a half-life of 1012 years at 25°C) allows the formation of very long polynucleotides that are remarkably stable despite the large number of phosphate ester bonds, an attribute that is essential for long-term storage of genetic information.
> …The energetics of phosphorylation are relatively well balanced, because the energy of the phosphate ester linkage is similar to that of the ATP β–γ anhydride bond. In fact, protein kinase equilibrium constants for the (protein + ATP → P.protein + ADP) reaction range from 2 to 50, and many protein kinases will readily work in reverse to dephosphorylate a phosphoprotein in the presence of ADP, generating ATP. Thus, in the cell, phosphorylation is dependent on the high ATP/ADP ratio, which prevents the back reaction. Although phosphate esters are rather stable chemically, they can be readily hydrolysed by an appropriate enzyme catalyst under physiological conditions. The relatively high energy of phosphate monoesters ensures that once they are hydrolysed, they cannot be re-formed through phosphorolysis by reaction with free phosphate, thus rendering dephosphorylation irreversible.
In chemical terms, the differentiation from the 20 unmodified amino acids in the charge and ionic/hydration shell size of phosphoamino acids leads to distinctive intra/intermolecular interactions (stronger and more stable H bonds/salt bridges than through Glu/Asp). PO43‒ is also particularly well-suited for interacting with arginine and lysine thanks to its bifurcated charge (apt for forming bifurcated rather than linear hydrogen bonds).
> Protein phosphates can act sterically or ionically to regulate function or the interaction of another protein or small molecule, or more commonly to elicit a conformational change within a protein monomer or an allosteric transition within a protein multimer.
By comparison, sulphates are only monoanionic at physiological pH, both pKa's ~2 (sitting in group 16 next to P in 15, sulphates have only two ionisable oxygens to phosphates' three). As such, not only are the energetics less favourable, but there's no basis for a distinction against the [unmodified] acidic amino acids to be exploited in signalling.
Hunter suggests arsenate (the next eligible group 15 member) as the most likely contender for a phosphate alternative. Indeed, a NASA research fellow notoriously claimed in a 2010 Science publication that microbes could resort to an 'arsenic-based life' when phosphorus was scarce. Such arsenate esters are however highly unstable in aqueous solution, with half lives below 0.02s, and As has prohibitively high reactivity with cysteine thiols (the basis for arsenic toxicity) to pose a stable signalling molecule.
Elsewhere in the PTRSB issue is a review of the evolution of the eukaryotic protein kinases as dynamic molecular switches, which notes that "eukaryotic protein kinases, in general, are not efficient enzymes, and efficient catalysis is not a requirement for a switch", in contrast to metabolic kinases such as hexokinase.
> The other distinction between EPKs and metabolic kinases is the amount of substrates they encounter in the cell. There are usually millimolar concentrations of small molecules that get phosphorylated by other kinases, but EPKs and their protein substrates are present in submicromolar or even nanomolar concentrations. This substrate concentration difference between metabolic kinases and EPKs is one major reason why metabolic enzymes have high Km values with efficient kcat values while EPKs have low Km values with relatively poor kcat values. Because of these drastic concentration differences, EPKs have evolved efficient mechanisms such as scaffolding, small protein: protein interaction domains and peptide-binding pockets in order to increase the effective concentrations of EPK–substrate encounter complexes.
Also in this issue: Design principles underpinning the regulatory diversity of protein kinases & Modular evolution of phosphorylation-based signalling systems.
Hunter also points to a classic Science paper by Frank Westheimer 25 years prior: Why nature chose phosphates. Westheimer questioned why such a poor leaving group was used, where organic chemists would not have.
> A possible explanation for the role of phosphates begins with a paper published in 1958 by Davis entitled "The importance of being ionized".(pdf) Davis's thesis was that living organisms must conserve their metabolites within the cell membrane. If these compounds had diffused through the membranes of evolutionarily primitive organisms, they would have been lost by dilution in the water outside the cell. Most electrically neutral molecules will have some solubility in lipid and will pass through a membrane; most ionized molecules, that is, salts, are insoluble in lipids. More precisely, the pK of an acid should be less than 4 and that of a base greater than 10 to ensure that only a small fraction of the compound remains in the un-ionized form at physiological pH. This general rule is not absolute. Polyhydroxylated compounds such as sugars may be lipophobic without being ionized, and compounds that are extremely insoluble in water, such as steroids, either become part of the membranes or are conserved within cells in special ways or both. But certainly molecules can be kept within membranes if they remain ionized. The first pK's of phosphoric acid and of phosphate mono- and diesters are about 2, so that phosphates are ionized at physiological pH and therefore are trapped within cells.
An electrostatic argument is used later to explain the chemical stability of the phosphate monoester bonds to proteins: that the ionised phosphate group would repel anionic nucleophiles.
> As explained above… metabolites should be charged to keep them within the cell membrane, and this charge must be negative, since a negative charge has an important second effect: it sharply diminishes the rate of nucleophilic attack on the ester. Nucleophiles, such as hydroxide ion, are repelled by negative charges and therefore react less rapidly with anions than with neutral substrates. Furthermore, the same effect is qualitatively present with respect to electrically neutral nucleophiles such as water. In this case, attack involves the pushing of a lone pair of electrons from the nucleophile into the electrophilic site of reaction.
> ### The effect of negative charge
> Two questions immediately arise. First, how much slower is nucleophilic attack on a negatively charged substrate as compared with attack on a neutral species, and second, would not this effect also occur with diesters of other tribasic acids such as citric acid? The answers to these questions come from standard physical-organic chemistry and show the advantage of phosphates.
> The effect of charge on ionization constants and on reaction rates was noted and explained by Bjerrum in 1923; a crude model that gives moderately good quantitative results was published in 1938. To what extent will a negative charge retard the approach of a nucleophile to a phosphorus or carbon atom, or indeed to the atom of any element, in an ester? Qualitatively, the effect will be comparable to the effect of a negative electric charge on the ionization of the corresponding polybasic acid. When a polybasic acid such as phosphoric acid or citric acid ionizes, the covalent bond between a proton and the oxygen atom to which it is attached must be broken, and the electrostatic work required to take the proton to infinity in the field ofthe negative charge of the incipient anion must be overcome. The work required to remove a second proton from a dibasic acid should be similar to that to remove the first proton, except that the second proton must be removed not only in the field of the incipient charge on the oxygen atom to which the proton was attached but also in the additional electrostatic field of the negative charge produced by the first ionization.
> …The rate for attack by hydroxide ion on trimethyl phosphate is less than that for attack on ethyl acetate by a factor of 25; the rate for attack of hydroxide on the anion of dimethyl phosphate is less than that for attack on ethyl acetate by a factor that approaches 5 million. (The statistical factors are sufficiently small by comparison with these numbers that they can be neglected.) The half-time for the hydrolysis of dimethyl phosphate in a solution of 1N alkali at 1 10°C is about a day (15). Such stability guarantees that genetic material will survive. Further questions related to hydrolysis are discussed later.
> ### Dianions of monoesters of phosphoric acid
> But another problem arises in connection with the chemistry of phosphates. The third ionization constant of phosphoric acid is smaller than the second by a factor of 105. Should not then the hydrolysis of the monoester dianions of phosphoric acid be slower than that of diesters by a similar factor? If dimethyl phosphate monoanion is hydrolyzed in LN base at 110°C with a half-time of a day, will the hydrolysis of the dianion under the same conditions require 3000 years? Since, however, the second pK of phosphoric acid is about equal to physiological pH, a considerable quantity of monoanion will be present, and subject to nucleophilic attack.
> …Despite some argument about details, the need for monomeric metaphosphate has been firmly established. The species has been identified in the gas phase and in tertiary butyl alcohol, but a vigorous (if friendly) debate has centered on whether the monomeric metaphosphate ion is ever truly free in water.
> In phosphorylations, PO3- is transferred, for example, from ATP to various substrates. Nucleophilic attack has been inferred because many such reactions result in stereochemical inversion at phosphorus; at present, only the solvolysis of p-nitrophenyl phosphate, made chiral at the phosphorus atom with 16O, 17O, and 18O, is known to proceed with racemization. Paradoxically, however, the kinetics for some reactions that proceed with inversion at phosphorus resemble those of SN1 processes.
> In the transition state for phosphorylations, metaphosphate [PO3‒] is essentially completely formed, while the bond to the monomeric metaphosphate residue from an incoming nucleophile has barely begun to form. In such a loose transition state, the monomeric metaphosphate ion is nearly, even if not completely, free. Protons are known only in the gas phase and are never free in water, yet mechanistic chemistry requires the concept of protons. The concept of monomeric metaphosphates is needed to explain how monoesters of phosphoric acid and ATP, for example, react in the absence of enzymes at measurable rates despite their negative charges, and react by enzymatic catalysis sufficiently rapidly to participate in metabolism.
Given that phosphate provides an imperfect leaving group, (de)phosphorylation at a speed significant enough for biological signalling is reliant on enzymatic catalysis. As such, there's no 'noise' in the signal from [uncatalysed] self-removal or self-modification. Spatial control from scaffolding proteins, subcellular localisation etc. also make it "robust" in the face of noise from other sources.
The question of whether metaphosphate is 'ever truly free' in solution relates to the mechanism of phosphorylation, which (perhaps surprisingly for such an important process) remains unsettled.
The general mechanism of kinases may be either 'associative' or 'dissociative' - proceeding via a metaphosphate intermediate or a pentacoordinated, metaphosphate-like transition state. Which is correct remains a matter of debate. If viewed as a single concerted process ("associative"), a nucleophile attacks the phosphate donor (invariably ATP) proceeding via an SN2 type reaction (with Walden inversion of chirality), through a pentacoordinated 'loose transition state'. A 'tight transition state' in this case would be one showing greater phosphorane characteristics, i.e. the P atom having more net bonding to the leaving group and nucleophile atoms than at the initial, 'ground' state (see visual explanations below, from the 2011 Annual Rev. Biochem. from Lassila et al.) pdf
A 2004 report in PNAS probed the rate of phosphorylation using kinetic isotope effects (KIE), for the prototypical small monomeric GTPase Ras, concluding in support of the loose transition state (thus SN2-type kinetics, though not exactly for 'associative' catalysis).





















